Sorry this is now out of date and Illumina keeps tweaking Basespace so rather than make another detailed guide that will be out of date soon, I’m keeping the old guide as reference and trying to update a quick guide more frequently.
MARS even quicker guide to downloading data from Basespace
Log into Basepace, navigate to Projects, click the box next to the project you want to download, above the list of projects there is a piece of paper icon, click that paper icon, clidk Download , click Project. You will be prompted to install the Basespace Download Manager, say yes, and your download should begin.
UConn MARS quick guide to getting data off BaseSpace
Yeah! The wet lab processing is done and you have data. You’ve gotten an email from basespace-noreply@illumina.com with a link to your project. Clicking that will take you to BaseSpace where you can log in and accept the invite in the pop-up that appears on screen. Another pop-up will appear in the bottom right corner of the screen to confirm the project share.
I prefer Basespace Classic which organizes data as “Samples” rather than “Biosamples”. To switch to Classic, click on your user name , then click “Settings”, toggle to “Classic”.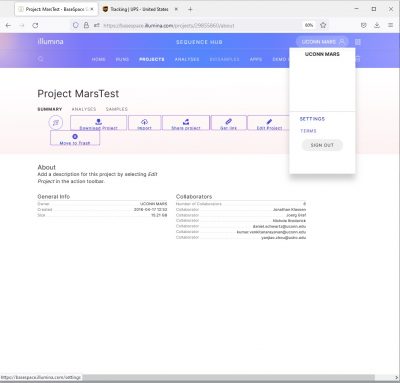
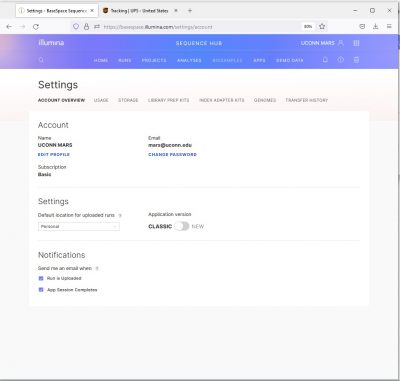
Once you have your Basespace set to Classic, click “Projects” at the top center of your screen.
Select the project that you want to work with.
Click “Samples” under the project title.
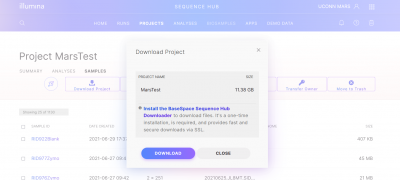
To download your raw data (1 forward and 1 reverse fastq for each sample), you can either use the “Download Project” button on the project screen, or you can select individual samples and then use the “Download Samples” button. If you are manually selecting samples, note that the “Sample ID” button only selects 25 samples (1 page) at a time.
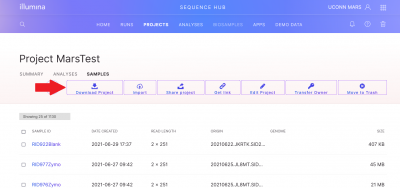
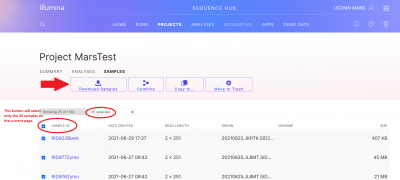
A download screen will pop up, if this is the first time you are downloading from BaseSpace you will need to Install the BaseSpace Sequence Hub Downloader. Then click Download your files.
All of the files from one project will go into a folder. Within the project folder, each sample will be in its own folder; this is where the two fastq’s will be. We use the sample barcode to identify each sample so the fastq are named with that barcode, but I tell BaseSpace to use your sample name as the folder name. BaseSpace adds 8 numbers to the end of the project and sample folders that you can just ignore.
If you are analyzing amplicons you might want to check out my mothur batch processing file or my bash file. The batch file has more details of whats happening, the bash file is what I’m actively using on the UConn/UCH HPC1 so there may be slight differences.
If you are analyzing microbial genomes, check out the Computational Biology Core’s tutorial.
If you don’t want to deal with the BaseSpace web interface, you can use BaseMount (a CLI from Illumina). There is a for loop at the top of my bash file for downloading all files for a project and renaming each file with your sample name instead of MARS’ unique identifier for the sample.
Feel free to email, call (860-486-1417), or stop by MARS if you have questions.
Happy Analyzing!!
Common issues:
If you set up your BaseSpace account with a different email than you use with me, this invite won’t work, but I can issue a new invite to whatever address you’d like me to use. If you click the link, log in, and get a message that the link is no longer valid just let me know and I’ll reissue the invite (occasional BaseSpace glitch).
**UPDATE** as of Nov 2016 Illumina changed this file structure and there are no longer “Data/Intensities/BaseCalls/” folders. If you have old and new samples sequenced by MARS, be aware that they will have different file structures.